
Login (User Login; top right of page) with your web user or complete this form to download.
Werum PAS-X와 손쉽게 통합되는 zenon MSI 인터페이스

페이지 내 다음 영역으로 이동하기:
제약 회사들은 생산 라인과 공정을 더욱 유연하게 만들고 데이터 무결성 규정을 준수해야 한다는 압박에 직면해 있습니다. 이러한 요구 사항을 충족하기 위해서는 기능뿐만 아니라 사용 편의성 측면에서도 제조 실행 시스템(MES)과 산업용 제어 소프트웨어가 필요합니다. 하지만 대다수의 기업은 여전히 오류 가능성이 높은 수기 문서에 의존하고 있습니다. 따라서 MES와 산업 제어 소프트웨어 간의 격차를 해소하는 것은 더욱 어려워지고 있습니다.
종이 기반 배치 기록에서 전자 기록의 전환은 생명과학 산업에서 경쟁력을 유지하는 데 가장 중요한 요인 중 하나입니다.
고전적인 접근법의 문제점 및 한계
오늘날의 접근 방식에서 MES는 배치 ID, 제품 이름, 레시피 이름과 같은 모든 파라미터를 포함한 하나의 메시지로 메시지를 교환하는 데 사용되는 반면, 대부분의 ICS 소프트웨어는 실시간 값(TAG)을 사용합니다.
모든 IT 시스템와 마찬가지로 MES 시스템도 SCADA 및 PLC와 실시간으로 값을 교환하도록 설계되지 않았습니다. MES와 현장 간의 통신에는 특정 순간에 메시지를 교환하는 트랜잭션 방식이 사용됩니다.
두 방식 간에는 변환이 쉽지 않아 데이터 무결성이 보장되는 데 한계가 있습니다. 생산 결과 및 GMP 예외(예: 크리티컬한 제품 품질 알람)가 MES 시스템으로 제대로 전송되지 않을 수 있기 때문입니다.
따라서 운영자는 필요한 유연성을 ㅎ하기 위해 산업 제어 소프트웨어와 간소화된 MES 통합이 필요합니다.
플랜트에서 일상적으로 발생하는 문제를 해결하기 위해서는 적합한 산업용 제어 소프트웨어가 필요합니다.
현재 OT와 IT 통합의 한계를 해결하기 위해 Werum PAS-X MSI 플러그 앤 프로듀스(메시지 기반 현장 통합)라는 통신 인터페이스가 등장했습니다.
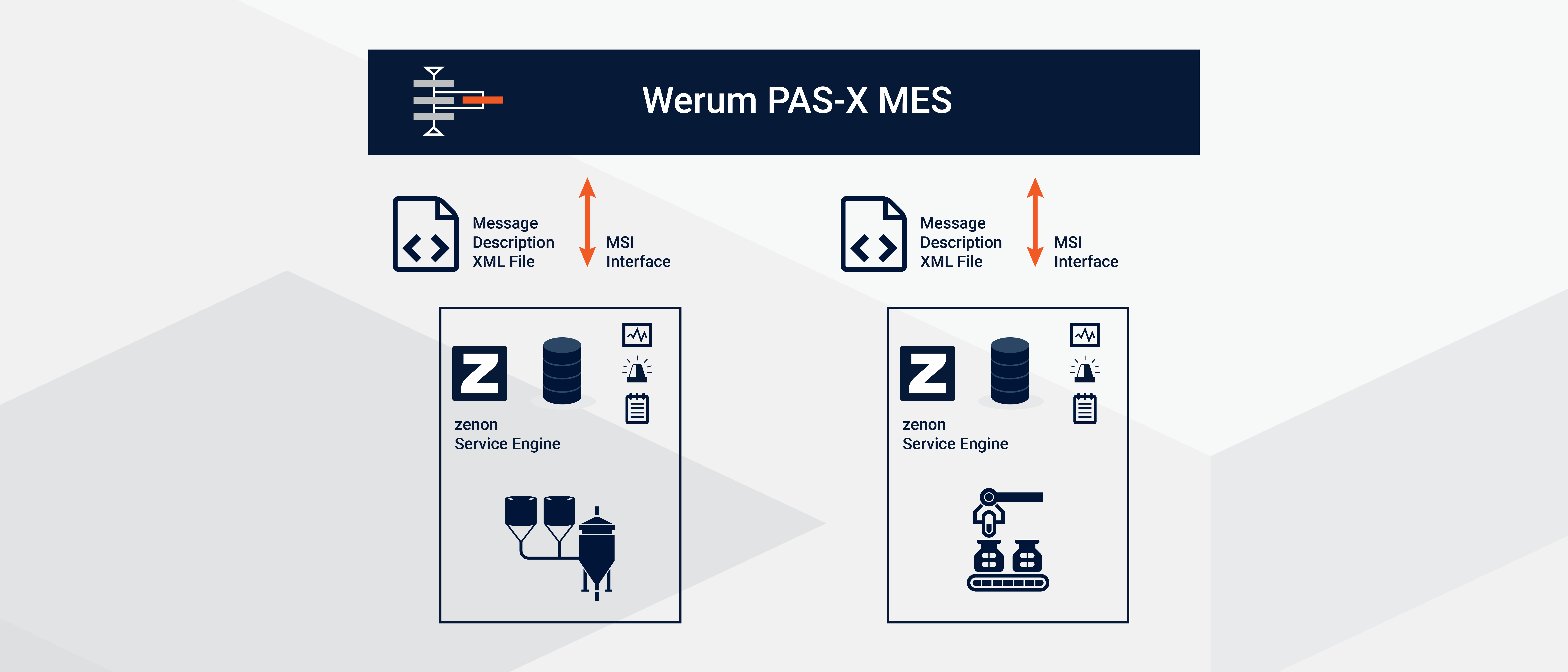
Körber가 개발한 Werum PAS-X MES는 제약, 바이오테크, 세포 및 유전자 치료 분야에서 전자 배치 기록(EBR: Electronic Batch Records)을 구축하는 용도로 가장 널리 사용되는 MES입니다. PAS-X MES는 MSI 어댑터를 통해 현장 장치와 통신합니다. 이 어댑터는 메시지 기반 솔루션으로 설계되었으며 업계 표준 통신 기술인 OPC-UA 및 REST 기반 웹 서비스를 완벽하게 지원합니다. PAS-X MES는 메시지 형태의 배치 파라미터를 산업용 제어 시스템에 전송하고 값이나 GMP 예외를 수신합니다.
구성 가능한 MSI 커넥터가 기본 내장된 zenon을 산업용 제어 시스템으로 이용하면 기계와 제조 라인을 손쉽게 연결하여 Werum PAS-X MES와의 격차를 좁힐 수 있습니다.
Werum PAS-X와 통합된 zenon
zenon의 MSI 인터페이스를 사용하면 PAS-X MES와 직접 배치 정보를 주고받고 장비로 전송할 수 있을 뿐만 아니라 GMP 알람 데이터를 MES로 전송할 수 있습니다. zenon PAS-X MES 통합을 통해 오류를 줄이고, 배치 출하 시간과 배치 간 다운타임도 줄일 수 있습니다. 하지만 가장 중요한 것은 현대 생명 과학 시설에 꼭 필요한 유연성을 제공한다는 점입니다.
현재 버전에는 새롭고 혁신적인 기능이 포함되어 있어 다음과 같은 MSI 메시지를 지원하는 탁월한 수준의 사용성을 보장합니다
- 오더 파라미터 메시지: PAS-X MES가 파라미터를 zenon에 전송하거나, zenon이 계측치와 값을 다시 전송하는 데 사용됩니다. (예: 배치 시작, 공정 중 제어, 배치 종료)
- 예외 메시지: 배치 실행 중 GMP와 관련된 편차가 발생했을 때 zenon에서 트리거됩니다. (예: 멸균 온도 하한 알람이 활성화된 경우).
- 오더 상태 메시지: 현재 장비 상태 또는 진행 중인 작업에 대해 PAS-X MES에 알리기 위해 zenon에서 트리거됩니다. (예: "PLC에 레시피 다운로드 진행 중 ...").
- 오더 취소 알림: 장비의 주문 또는 레시피가 중단되었음을 PAS-X MES에 알리기 위해 zenon이 트리거합니다.
제약 생산 라인의 생산성과 유연성에 대한 높은 요구사항을 충족시키기 위해 zenon PAS-X MES 통합은 운영에 필요한 기능을 제공합니다.

팩트 시트 다운로드하기
zenon PAS-X MSI 플러그 & 프로덕션 인터페이스 - MES와 생산 간의 직접 통신
오래된 시스템을 최신 환경에 통합
이 시나리오는 새로운 기계에 대한 '플러그 앤 프로듀스' 접근 방식을 제공합니다. 하지만 회사가 기존부터 보유해온 기계도 등한시하면 안 됩니다. 오래 전 설치되어 문제 없이 기능해왔지만 Werum PAS-X MES나 통합 데이터 스토리지 기능으로 직결되는 인터페이스가 없는 기계는 어떻게 해야 할까요? 이런 경우에는 기존 기계와 MES 시스템을 잇는 미들웨어가 필요합니다.
zenon 소프트웨어 플랫폼을 사용하면 확장성이 뛰어난 AIL(Automation Integration Layer)이라고 하는 모듈식 미들웨어를 구축할 수 있습니다. 이 구성에서 zenon은 광범위한 PLC(Simatic S5 등 레거시 PLC 포함!) 및 현장 장치를 연결하고, 수집된 데이터를 맥락화하여 히스토리안을 구축하고, MSI 인터페이스를 통해 Werum PAS-X MES와 통신하여 기존 기계를 모두 포함한 통합 환경을 제공합니다.
예를 들면, zenon이 PAS-X MES로부터 배치 개시 메시지를 수신합니다. zenon이 수신된 메시지를 해석하고, 수신자 기계에 장착된 기존 컨트롤러가 인식 가능한 형태로 정보를 매핑하여 라우팅합니다. 배치 맥락화가 기계 내에 없더라도 zenon AIL에는 구현되어 있으므로 계측치나 GMP 알람 등 수집된 정보가 배치 종료 시점에 MES 시스템으로 올바르게 전송됩니다.
zenon은 Werum PAS-X MES 외에도 히스토리안이나 ERP 등의 IT 시스템과도 통합 가능한 인터페이스를 갖추고 있습니다. 그리고 zenon은 수집된 데이터를 사용해서 그림 1과 같이 다양한 부서에 맞는 대시보드 또는 보고서를 생성할 수도 있습니다.

최신 환경 시나리오: PAS-X MSI 플러그 앤 프로듀스 지원 기계
전형적인 최신 환경 사용 사례는 zenon을 기계에 장착된 HMI처럼 사용하는 것입니다. 이 경우 클래식한 배치 개시 및 종료 명령 전달, 중요한 GMP 알람 공유를 위해 제조업체가 사전 구성한 Werum PAS-X MSI 인터페이스가 포함될 수 있습니다. MES와 교환할 메시지는 최종 고객과 사전에 논의하여 정할 수 있습니다. 기계는 MSI 인터페이스가 사전 구성된 상태로 고객에게 납품됩니다. 최종 고객의 PAS-X MES 시스템과의 통합을 위해서는 제조사가 준비한 MSI 메시지 정의를 공유하는 것만으로도 충분합니다. 마지으로 몇 가지 주소만 구성하면 기계와 MES 시스템 간의 통신이 수립됩니다. 기계 제조사가 준비한 메시지는 향후 최종 고객이 메뉴를 통해 GAMP SW CAT.4에 따라 구성을 변경하여 사용할 수 있습니다. 비용이 많이 드는 계층적 통합이 필요하지 않은 간편한 접근 방식입니다. 이러한 기계를 PAS-X MSI Plug&Produce Ready라고 부를 수 있는 이유입니다.

zenon의 PAS-X MSI 플러그 앤 프로듀스 지원 인증에 대한 내용은 Körber 보도자료에서도 확인하실 수 있습니다.
생명과학 산업을 위한 zenon MES 통합
바이오테크 MES와 제약 MES 모두에 zenon은 완벽한 연결성과 프로젝트 검증을 자랑하는 솔루션입니다. COPA-DATA의 zenon은 효율성, 비용, 데이터 관리의 이점을 제공하는 강력한 모듈형 플랫폼으로 라인 실행 시스템(LES: Line Execution System), ISA 88 모듈식 공정 자동화, 무중단 제조 등의 데이터 무결성 도구 제공합니다.
COPA-DATA는 산업의 변화에 맞춰 산업용 제어 소프트웨어를 지속적으로 개선합니다. 귀사의 비즈니스의 지속적인 발전을 위해 필요한 모든 로직과 알고리즘을 활용합니다. zenon과 zenon이 비즈니스에서 어떤 역할을 수행할 수 있는지 자세히 알아보려면 지금 문의 바랍니다.
